Gene Mutations
Wednesday, July 12, 2023
GENE MUTATIONS: RELEVANCE IN CANCER
“A recent analysis of 20,000 protein-coding genes derived from 3284 cancers revealed a total of 294,881 mutations of which only 125 mutated driver genes were identified comprising 71 suppressor genes and 54 oncogenes.”
Although subject to broad interpretation, the mutational theory of cancer acknowledges that cancer is an evolutionary process and proposes that cancer cells achieve an adaptive advantage in their ecosystem as a result of one or more mutations affecting cell fate (the balance between cell division and differentiation), cell survival (allowing for growth advantage in an environment less supportive of normal cells, e.g., lower oxygen concentration, less availability of nutrients, higher acidity), or genome maintenance (primarily, impairment of effective DNA repair). Mutations that have the capacity to provide such an adaptive advantage are called “driver mutations”. Even though each driver mutation may result in only a 0.4% growth advantage (the difference between cell birth and cell death), a one-centimeter diameter cancer (weighing one gram) contains approximately one billion cells with an estimated 10-10 mutations/base pair/replication. Given the obvious limitations of translating from bacterial (E. coli) to human DNA mutation kinetics, it is still instructive to look at the differences in the rate of accumulation of mutations versus the rate of achieving adaptive fitness (see figure 1). Note that with a linear rate of mutational based genomic evolution there is a rapid increase in adaptive advantage followed by a slower, linear rate of change.
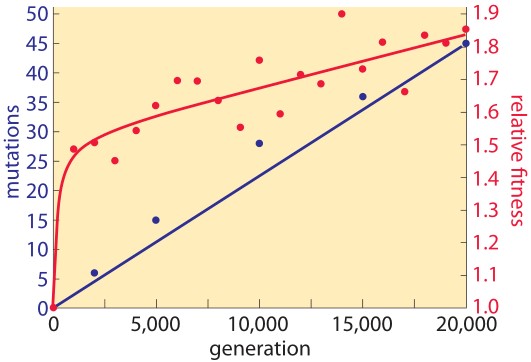
Figure 1. Rate of genomic evolution and relative fitness.
In human cancers, the number of non-synonymous mutations (those that alter the encoded amino acid sequence of a protein) ranges from approximately 200 in melanoma and lung cancer (cancers specifically associated with potent mutagens) to 9.6 in pediatric cancer (see figure 2). Given the relatively constant rate of change of mutation in normal cells accompanying cell division, it is important to note that the vast majority of mutations that occur are synonymous, do not alter the amino acid sequence in any protein, and are thus functionally invisible. Of the non-synonymous mutations, once again, the majority have no effect on carcinogenesis and are called “passenger mutations”. Essentially, they are there for the ride. Two key, initiating types of driver mutations are 1) gatekeeper mutations, which regulate cell proliferation and thus provide a growth advantage, and 2) caretaker mutations, which protect the integrity of the DNA through their role in repair. Examples of the former are RB1, p53, and APC; of the latter, p53 (with dual functionality), and BRCA1.
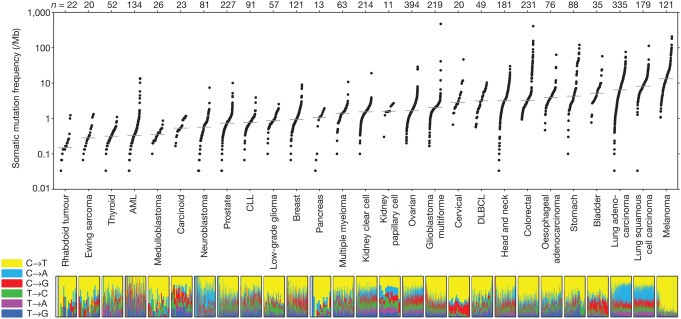
Figure 2: Number of somatic mutations in representative cancers
“Mary Crowley Cancer Research routinely evaluates cancer tissue from patients in order to identify actionable mutations through collaborations with institutions capable of performing whole-genome and/or whole-exome sequencing…”
A recent analysis of 20,000 protein-coding genes derived from 3,284 cancers revealed a total of 294,881 mutations, of which only 125 mutated driver genes were identified (using specific parameters), comprising 71 suppressor genes and 54 oncogenes. To add to the complexity, in addition to the role of driver genes, phenotypes derived from passenger mutations can undergo exaptation, by which their function shifts due to changes in evolutionary pressures, resulting in cancer supportive activity. Mary Crowley Cancer Research routinely evaluates cancer tissue from patients in order to identify actionable mutations through collaborations with institutions capable of performing whole-genome and/or whole-exome sequencing to discover point mutations, insertions and deletions (indels), copy number alterations, and gene fusions (see figure 3). The turnaround time continues to improve to allow for more rapid application of results to choice of therapeutic options.
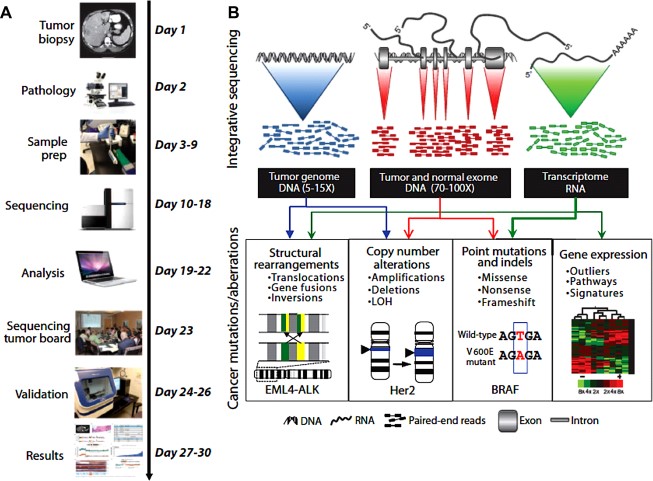
Figure 3: Integrative clinical tumor screening
“The EWS/FLI1 fusion is present in the tumors of eighty five percent of Ewing’s sarcoma patients 60% of which are type I.”
An exciting example of the application of this technology at Mary Crowley is a new protocol being exclusively developed for the clinic, Phase I Trial of pbi-shRNA™ EWS/FLI1 Type 1 lipoplex (LPX) in Subjects with Advanced Ewing’s sarcoma. The study will assess the safety and effectiveness of an innovative approach, RNA interference, in blocking the expression of a driver fusion gene, EWS-FLI1, integral to Ewing’s sarcoma, a childhood cancer. Importantly, this fusion gene has heretofore been undruggable, i.e., not responsive to currently available therapeutics. Only patients with the type I fusion, defined by a specific site at which the EWS gene fragment connects with the FLI1 fragment, will be candidates. The EWS/FLI1 fusion is present in the tumors of 85 percent of Ewing’s sarcoma patients, 60 percent of which are type I. Next generation sequencing (NGS) will enable the selective application of this therapy to only those patients capable of benefiting from it should it prove to be effective.
How effective is genomic sequencing in directing cancer therapy? In a recent prospective multi-institutional study of the role of NGS, the outcomes of 122 patients with a variety of cancer types receiving matched Phase I therapy (that directly impacted the identified target) were compared with the outcomes of 66 patients receiving unmatched therapy. Using matching scores, high scores were independently and significantly associated with longer time to treatment failure and overall survival. These results are impressive given the limitations of the study: the possibility of less than optimal accession of the target, poor pharmacokinetics, and a range of dosing levels presumably including some that were suboptimal. In any event, integrative clinical genomic tumor analysis is a major step forward toward the control and eventual cure of cancer.

Related Posts
Discover more posts like this.