Cancer Pathways
Thursday, July 20, 2023
Michael Foster
Molecular Data Analysis: Blocking Specific Pathways
There are eight hallmark biological capabilities necessary for cancer development and persistence: (1) sustained proliferation, (2) evasion of growth suppressors, (3) death resistance, (4) replicate immortality, (5) angiogenesis, (6) invasion ± metastability, (7) reprogrammed energy metabolism, and (8) immune evasion. These capabilities support one or more of three core processes, i.e., cell survival, cell fate and genome maintenance. These core processes are sub served by one or more of 12 signaling pathways.
Blocking the cancer specific relevant pathway[s] underlying the core processes will cripple cancer persistence and progression (Figure 1).
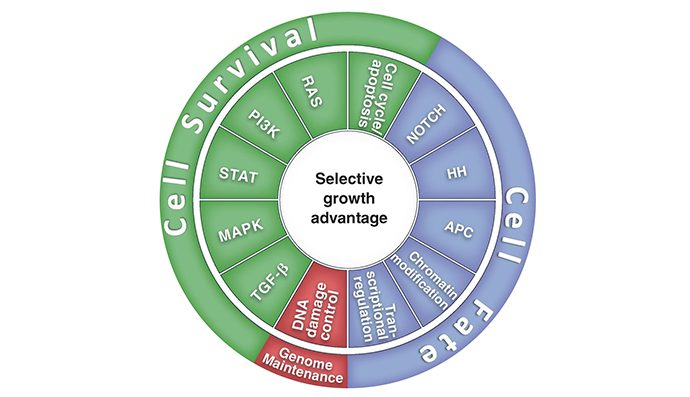
Figure 1. Twelve signaling pathways, which are responsible for 3 cancer core functions involving cell survival, fate, and genome maintenance.
Fortunately, the multigenomic/proteomic components of each of the relevant pathways are dependent on a limited number of aberrant and rewired “hub” elements comprising “driver” genes (oncogenes and suppressor genes); rate-limiting genes/proteins, and high-information transfer genes/proteins. The targeting of these drive genes is feasible and can effectively block the cancer-specific relevant pathways. Examples of specific “target the target” benefits that have emerged from preclinical and clinical molecular targeting studies are B- vemurafenib/RafV600E, Tagrisso/EGFRT790M, crizotinib/ALK, and Entrectinib/NTR1. The demonstration of dramatic responses, PFS (progression free survival) and OS (overall survival) benefits in numerous NDA applications has sufficed to justify FDA approval of an increasing number of targeting therapeutics.
As a case in point, the PAM pathway is a complex signaling network component with multiple negative feedback systems and a variety of mechanisms of development of adaptive resistance (see Figure 2).
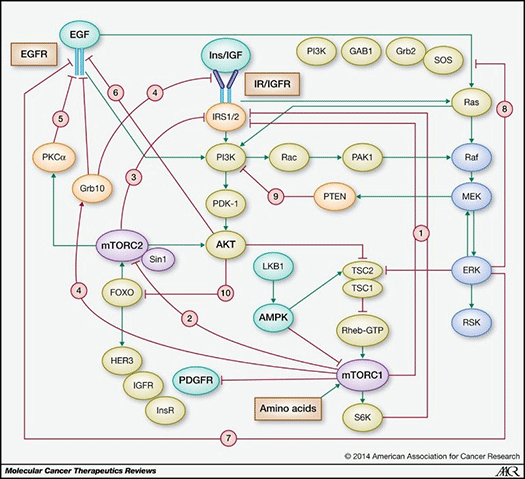
Figure 2. PAM pathway (stimulatory connections in green; negative feedback loops in red) (from Rozengurt E et al, 2014).
Chronic inhibition of the PAM downstream node mTORC1 relieves redundant negative feedback loops on PI3K/AKT via IRS-1, thereby overriding pathway interference (see Figure 3) that, however, can be subverted by upstream element inhibition, e.g., PI3k/AKT. That p70S6K, a substrate of mTORC1, is amplified in 8.8% of primary breast cancer and overexpressed at the mRNA level in 38% of breast cancers may allow for mTORC1 independent expression, further supporting the rationale for the use of a dual AKT/p70S6K moiety. In terms of mechanism, p70S6 kinase plays a critical role integrating the HER-family and PI3k pathways. Its activation is associated with resistance to both trastuzumab and lapatinib. In addition, overexpression of RSK3 (ribosomal S6 kinase, p90RSK3) has also been shown to mediate resistance to PI3k/mTOR inhibition and is associated with resistance to lapatinib. A Phase I dual AKT/p70S6K targeting therapeutic has been shown to have in vitro potency against RSK3 although to a lesser degree than to S6k and AKT 1,3 (private communication).
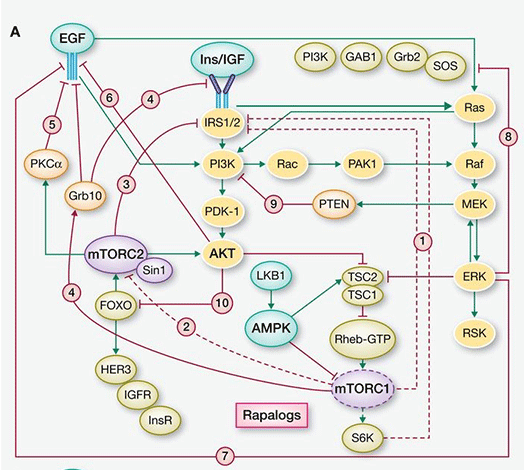
Figure 3. mTOR inhibition of the PAM pathway (stimulatory connections in green, negative feedback loops in red, pathways activated by suppression of negative feedback loops highlighted in yellow) (from Rozengurt et al, 2014).
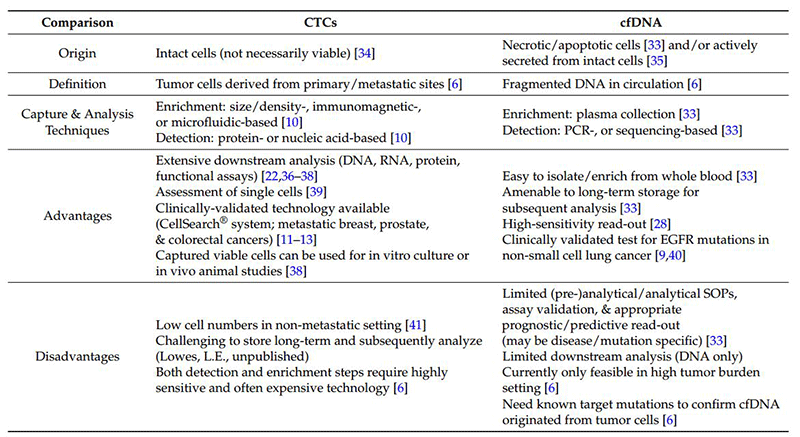
However, even with a dual PI3k/mTORC1, 3 inhibitor, there are remaining avenues for resistance to emerge via suppression of multiple negative feedback loops resulting in over-activation of the MEK/ERK pathway via a PI3k-independent loop, as well as FOXO-mediated up-regulation of tyrosine kinase receptors (e.g., HER3, IGFR, and InsR) (see Figure 4). Knowledge of these compensatory pathways informing the likely modes of development of adaptive resistance offers the opportunity for rapid assessment of tumor-specific genomic/proteomic mechanisms of resistance at time of progression. With such, this allows for the institution of mechanism-specific therapeutic combinations to circumvent resistance, e.g., a dual PI3k/mTORC1,3 inhibitor + MEK inhibitor, a dual PI3k/mTORC1,3 inhibitor + AZ8931 (EGFR/HER2/HER3 inhibitor, or a dual PI3k/mTORC1,3 inhibitor + IGFR/InsR inhibitor). Further, considering the continuing expansion of CTC (circulating tumor cell) detection and isolation techniques (e.g., microfluidic), both proteomic and genomic (next-generation sequencing (NGS), digital droplet PCR) analyses have the potential for detection of emerging pockets of resistance prior to clinical progression, increased frequency of assessment (especially in difficult to biopsy areas), and a broader view of heterogeneic tumor cell biology. Likewise, although limited to DNA analysis, cfDNA (circulating free DNA) would allow for genomic interrogation to identify resistance mediators, such as KRAS mutations and, possibly, some activating mutations of AKT.
Figure 4. Dual PI3k/mTOR inhibition of the PAM pathway (stimulatory connections in green, negative feedback loops in red, pathways activated by suppression of negative feedback loops highlighted in yellow (from Rozengurt E et al, 2014).
The following is a summary comparison of CTC and cfDNA.
In conclusion, cancers evolve, as do all emergent processes. The traditional reductionist approach to therapeutic intervention will not suffice. At the Mary Crowley Cancer Center, in collaboration with each patient and their referring physician, interrogation of the molecular fingerprint of each patient’s cancer followed by a system analysis of its integrated components has long been our approach to personalized (aka precision) therapy.
Blocking Cancer Pathways: PERSONALIZED MEDICINE AT MARY CROWLEY
“Almost all gene modifications can be classified in one or more of 12 signaling pathways.”
In February 2001, the Human Genome Project published the 90 percent complete sequence of the three billion base pairs in the human genome containing approximately 30,000 genes. As a result of advances in sequencing technology (whole genome and whole exome), commercialization permitting wide-ranging access, and improved turn-around time, 138 mutated cancer driver genes have been identified to date. These cancer driver genes are defined as mutated genes that provide tumor cells with a selective growth advantage in one or more of the local, regional, and systemic environments. There are a significantly larger number of mutations that, although present in the cancer cells, occurred during the preneoplastic normal growth phase and, insofar as these mutations did not initially provide an adaptive advantage, are called passenger genes. Yet, these mutated genes can undergo exaptation that ex post facto describes the shift in their function due to changes in evolutionary pressures as a result of the functional rewiring. These changes are facilitated by the driver genes that result in a shift from neutral to cancer supportive activity. Additional perturbations in gene functionality can also be due to non-mutational changes in gene copy number, insertions and deletions (indels), fusions and epigenetic modifications (heritable non-structural changes primarily due to changes in methylation). Data encoded in the DNA, essentially a template, is transcribed into messenger RNA (mRNA), a bio-molecular mold, then translated into proteins (the product) which influence the reaction rates of metabolites (catalysis) (see figure 1). All of these –omic levels (genomic-transcriptomic (mRNA)-proteomic-metabolomic) can interact with and modify each other. To make matters more complicated, mRNA can undergo alternative splicing by means of which encoding sequences (exons) can be rearranged, resulting in more than one mRNA per gene, producing protein isoforms with different, even opposing, functions. Furthermore, differences in protein abundance can only be attributed to mRNA levels in 20-40% of cases.
“In February 2001, the Human Genome Project published the 90 percent complete sequence of the three billion base pairs in the human genome containing approximately 30,000 genes…”
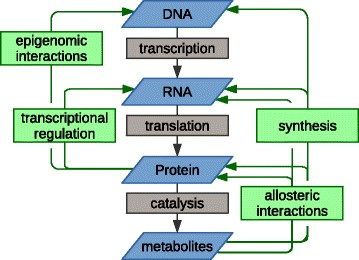
Figure 1: The multilevel relationships between –omic levels
At first, the array and abundance of infrastructural changes that confer a selective growth advantage to cancer cells by enhancing adaptive fitness can seem staggering and almost beyond our ability to choose which one[s] to target. However, there is an underlying order to this diversity. Almost all of these modifications can be classified into one or more of 12 signaling pathways (see figure 2), which subserve one or more of the 10 hallmark functions necessary for cancer growth and progression identified by Hanahan and Weinberg (see figure 3).
Figure 2: Cancer cell signaling pathways
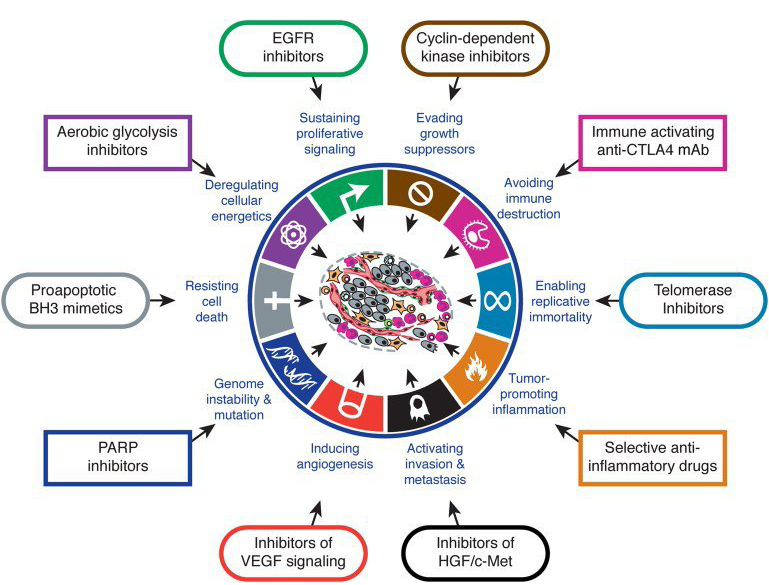
Figure 3: Hallmarks of cancer: capabilities necessary for cancer cell growth and progression.
“Almost all of these modifications can be classified into one or more of 12 signaling pathways, which subserve one or more of the 10 hallmark functions necessary for cancer growth and progression identified by Hanahan and Weinberg.”
The components comprising these 12 pathways interact with each other in a complex network made up of gene/protein nodes and connecting links, some of which are stimulatory and some inhibitory. The importance of any of these nodes to the operational efficiency of the network and, inversely, to error intolerance (the ability of the network to withstand one or more targeted damages to the nodes) is dependent on that node’s connectivity (the number of linkages to other nodes) as well as to which nodes it is connected. The linked nodes carrying high amounts of information between different clusters of nodes that are functionally distinct from each other are called “bottleneck nodes.”
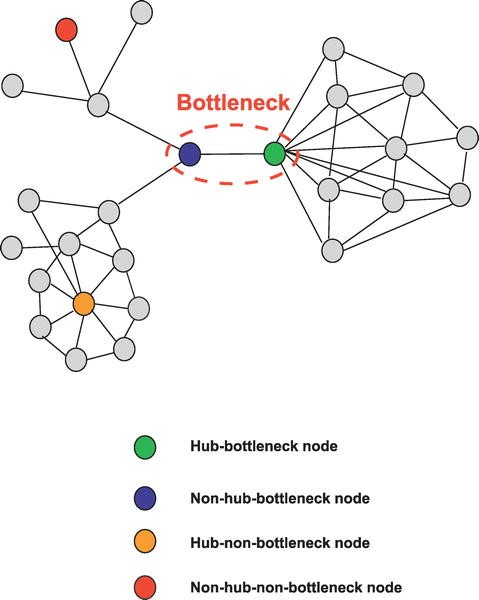
Figure 4: High information transfer “bottleneck nodes”.
These key ‘bottleneck nodes’ are those genes/proteins that are essential to network function which, in turn, are necessary for maintenance of the malignant process. Looking at this in another way, these nodes are the most fragile component of the network and, therefore, the most vulnerable sites within the functional network comprising the cancer—appropriate targets for therapeutic attack (see figure 5).

Figure 5: Targeting “bottleneck” verses “non-bottleneck” nodes
“These key “bottleneck nodes” are those genes/proteins that are essential to network function which, in turn, are necessary for maintenance of the malignant process.”
Understanding Cancer Pathways
Insofar as a number of different genes/proteins can have rate limiting effects in a dominant signaling pathway, systems analysis can allow optimization of limited target pathway disruption. Further, given the cross-talk between pathways and that the robustness of the cancer network involves alternative and/or compensatory secondary pathway utilization, it is likely that more than one pathway needs to be targeted. For example, in non-small-cell lung cancer with an EGFR inhibitor sensitizing L858R mutation in exon 21, effective targeted therapy can be bypassed by upregulation of the cMet receptor and its downstream pathway. Mary Crowley has continued to offer a number of protocols with multiple node targeting—a major step in the development of Personalized Medicine.

Related Posts
Discover more posts like this.